Research |
Global atmospheric chemistry
|
Understanding the global-scale dynamics of the chemical composition of our atmosphere is essential for addressing a wide range of environmental issues from air quality to climate change. Understanding this phenomenon enables us to evaluate and devise appropriate environmental policies, such as the Kyoto Protocol on global greenhouse gases emissions. Numerical modeling of global atmospheric chemical dynamics presents an enormous challenge associated with simulating hundreds of chemical species with time scales varying from milliseconds to years.
In my research, I have worked on the implementation of computationally efficient algorithms for calculating the time evolution of the concentration of chemical species in global 3-D models of atmospheric chemistry. I have also investigated the efficacy of adjoint based inverse modeling techniques for source attribution problems. Assessing model errors and uncertainties: I am very interested in understanding the large dependence of the results of Global Atmospheric Chemistry Models on spatial resolution, time stepping, and spatial averaging of meteorological fields, and the impact of these on the contaminant plume propagation. |

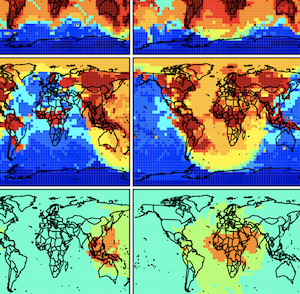
Percentage of fast species in the GEOS-Chem chemical mechanism at different altitudes using a threshold of d 1⁄4 102 molecules cm 3 s 1. White boxes in the bottom right panel are in the stratosphere. Results are for July 8, 2004 at 00 GMT. The full GEOS-Chem chemical mechanism includes 111 species to describe tropospheric ozone-NOx-VOC- aerosol chemistry.
From: M. Santillana, P. Le Sager, D. J. Jacob, and M. P. Brenner. An adaptive reduction algorithm for efficient chemical calculations in global atmospheric chemistry models. Atmospheric Environment. Volume 44, Issue 35, pp 4426-4431, Nov 2010. (PDF)) |
Related publications
|
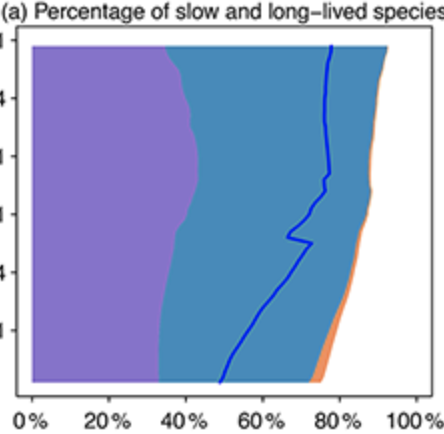
A machine-learning-guided adaptive algorithm to reduce the computational cost of integrating kinetics in global atmospheric chemistry models: application to GEOS-Chem versions 12.0.0 and 12.9.1
L Shen, DJ Jacob, M Santillana, K Bates, J Zhuang, W Chen Geoscientific Model Development 15 (4), 1677-16872022 Abstract
Global modeling of atmospheric chemistry is a great computational challenge because of the cost of integrating the kinetic equations for chemical mechanisms with typically over 100 coupled species. Here we present an adaptive algorithm to ease this computational bottleneck with no significant loss in accuracy and apply it to the GEOS-Chem global 3-D model for tropospheric and stratospheric chemistry (228 species, 724 reactions). Our approach is inspired by unsupervised machine learning clustering techniques and traditional asymptotic analysis ideas. We locally define species in the mechanism as fast or slow on the basis of their total production and loss rates, and we solve the coupled kinetic system only for the fast species assembled in a submechanism of the full mechanism. To avoid computational overhead, we first partition the species from the full mechanism into 13 blocks, using a machine learning approach that analyzes the chemical linkages between species and their correlated presence as fast or slow in the global model domain. Building on these blocks, we then preselect 20 submechanisms, as defined by unique assemblages of the species blocks, and then pick locally and on the fly which submechanism to use in the model based on local chemical conditions. In each submechanism, we isolate slow species and slow reactions from the coupled system of fast species to be solved. Because many species in the full mechanism are important only in source regions, we find that we can reduce the effective size of the mechanism by 70 % globally without sacrificing complexity where/when it is needed. The computational cost of the chemical integration decreases by 50 % with relative biases smaller than 2 % for important species over 8-year simulations. Changes to the full mechanism including the addition of new species can be accommodated by adding these species to the relevant blocks without having to reconstruct the suite of submechanisms. |
|
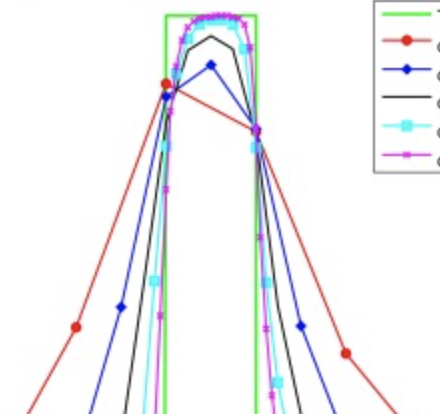
An adaptive method for speeding up the numerical integration of chemical mechanisms in atmospheric chemistry models
Shen L, Jacob DJ, Santillana M, Wang X, Chen W. Geoscientific Model Development . 2020;13 :2475–2486. Abstract
The major computational bottleneck in atmospheric chemistry models is the numerical integration of the stiff coupled system of kinetic equations describing the chemical evolution of the system as defined by the model chemical mechanism (typically over 100 coupled species). We present an adaptive method to greatly reduce the computational cost of that numerical integration in global 3-D models while maintaining high accuracy. Most of the atmosphere does not in fact require solving for the full chemical complexity of the mechanism, so considerable simplification is possible if one can recognize the dynamic continuum of chemical complexity required across the atmospheric domain. We do this by constructing a limited set of reduced chemical mechanisms (chemical regimes) to cover the range of atmospheric conditions and then pick locally and on the fly which mechanism to use for a given grid box and time step on the basis of computed production and loss rates for individual species. Application to the GEOS-Chem global 3-D model for oxidant–aerosol chemistry in the troposphere and stratosphere (full mechanism of 228 species) is presented. We show that 20 chemical regimes can largely encompass the range of conditions encountered in the model. Results from a 2-year GEOS-Chem simulation shows that our method can reduce the computational cost of chemical integration by 30 %–40 % while maintaining accuracy better than 1 % and with no error growth. Our method retains the full complexity of the original chemical mechanism where it is needed, provides the same model output diagnostics (species production and loss rates, reaction rates) as the full mechanism, and can accommodate changes in the chemical mechanism or in model resolution without having to reconstruct the chemical regimes. |
|
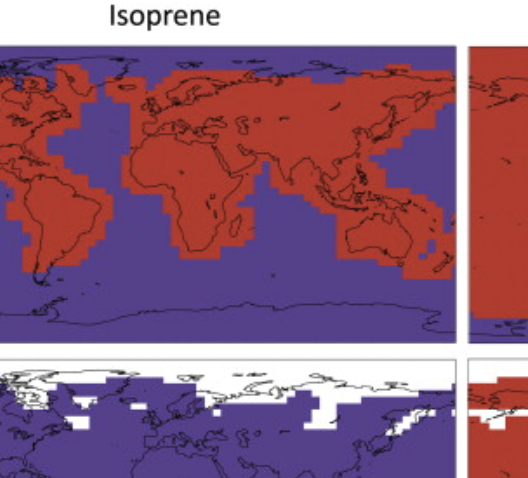
Estimating numerical errors due to operator splitting in global atmospheric chemistry models: Transport and chemistry
Santillana M, Zhang L, Yantosca R. Journal of Computational Physics. 2016;305 :372–386. Abstract
We present upper bounds for the numerical errors introduced when using operator splitting methods to integrate transport and non-linear chemistry processes in global chemical transport models (CTM). We show that (a) operator splitting strategies that evaluate the stiff non-linear chemistry operator at the end of the time step are more accurate, and (b) the results of numerical simulations that use different operator splitting strategies differ by at most 10%, in a prototype one-dimensional non-linear chemistry–transport model. We find similar upper bounds in operator splitting numerical errors in global CTM simulations. |
|
An adaptive reduction algorithm for efficient chemical calculations in global atmospheric chemistry models
Santillana M, Le Sager P, Jacob DJ, Brenner MP. Atmospheric Environment. 2010;44 (35) :4426–4431. Abstract
We present a computationally efficient adaptive method for calculating the time evolution of the concentrations of chemical species in global 3-D models of atmospheric chemistry. Our strategy consists of partitioning the computational domain into fast and slow regions for each chemical species at every time step. In each grid box, we group the fast species and solve for their concentration in a coupled fashion. Concentrations of the slow species are calculated using a simple semi-implicit formula. Separation of species between fast and slow is done on the fly based on their local production and loss rates. This allows for example to exclude short-lived volatile organic compounds (VOCs) and their oxidation products from chemical calculations in the remote troposphere where their concentrations are negligible, letting the simulation determine the exclusion domain and allowing species to drop out individually from the coupled chemical calculation as their production/loss rates decline. We applied our method to a 1-year simulation of global tropospheric ozone-NOx-VOC-aerosol chemistry using the GEOS-Chem model. Results show a 50% improvement in computational performance for the chemical solver, with no significant added error. |
|
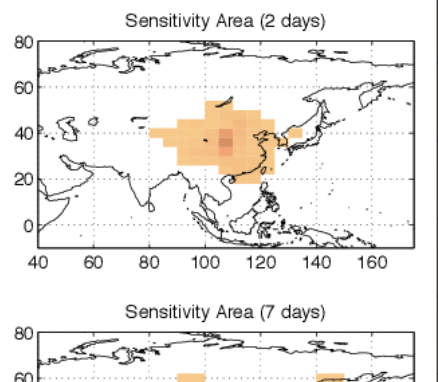
Quantifying the loss of information in source attribution problems using the adjoint method in global models of atmospheric chemical transport
Santillana M. arXiv preprint arXiv:1311.6315. 2013. Abstract
It is of crucial importance to be able to identify the location of atmospheric pollution sources in our planet. Global models of atmospheric transport in combination with diverse Earth observing systems are a natural choice to achieve this goal. It is shown that the ability to successfully reconstruct the location and magnitude of an instantaneous source in global chemical transport models (CTMs) decreases rapidly as a function of the time interval between the pollution release and the observation time. A simple way to quantitatively characterize this phenomenon is proposed based on the effective -undesired- numerical diffusion present in current Eulerian CTMs and verified using idealized numerical experiments. The approach presented consists of using the adjoint-based optimization method in a state-of-the-art CTM, GEOS-Chem, to reconstruct the location and magnitude of a realistic pollution plume for multiple time scales. The findings obtained from these numerical experiments suggest a time scale of 2 days after which the accuracy of the adjoint-based optimization methodology is compromised considerably in current global CTMs. In conjunction with the mean atmospheric velocity, the aforementioned time scale leads to an estimate of a length scale of about 1700km, downwind from the source, beyond which measurements, in conjunction with current global CTMs, may not be successfully utilized to reconstruct continuous-in-time sources. The approach presented here can be utilized to characterize the capabilities and limitations of adjoint-based optimization inversions in other regional and global Eulerian CTMs. |
